CIMZIA® demonstrated long-term broad spectrum efficacy across indications5-22
Image
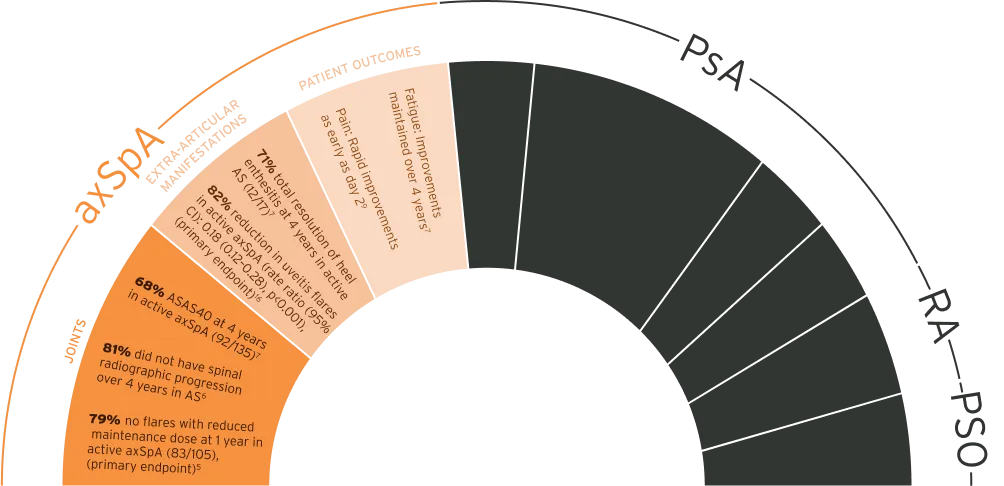
The safety and efficacy of CIMZIA® was assessed in patients with axSpA, PsA, PSO, and RA. In axSpA, CIMZIA® demonstrated efficacy on joints, extra-articular manifestations, skin, and monotherapy, as shown in the RAPID-PsAd phase 3 study. In RA, CIMZIA® demonstrated durable efficacy in the RAPID 1e, FAST4WARDf, and study 014g phase 3 studies. In PSO, CIMZIA® demonstrated efficacy in the CIMPASI-1h, CIMPASI-2h, and CIMPACTi phase 3 studies.5-22
Joints
C-OPTIMISE
CIMZIA® demonstrated sustained remission at a reduced maintenance dose in active axSpA5
Image
Image
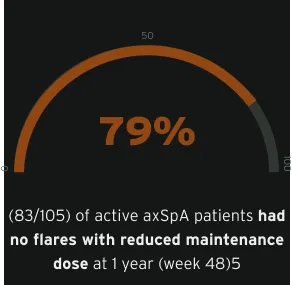
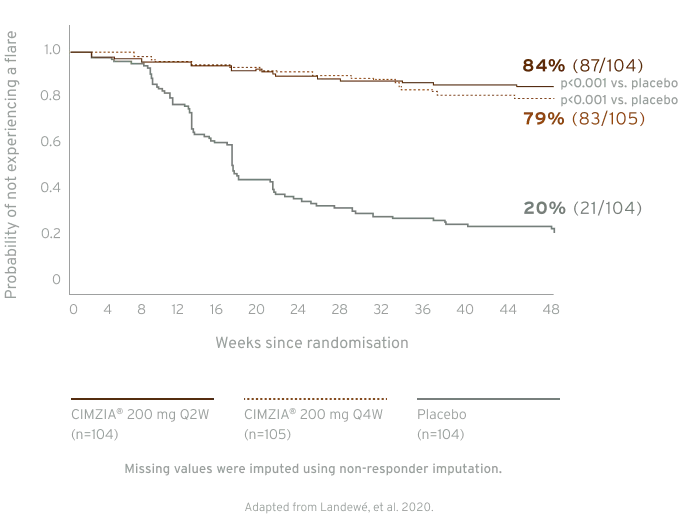
In the C-OPTIMISE study, the primary endpoint was met. The primary endpoint was remaining flare-free during the double-blind period. A statistically significant proportion of patients who continued on, either a full or reduced CIMZIA® maintenance dose, remained flare-free during the double-blind period between week 48 and 96, 83.7% (87/104), and 79.0% (83/105), respectively vs. patients who had CIMZIA® treatment withdrawn (placebo), 20.2% (21/104), p<0.001 vs. placebo in both groups.5
RAPID-axSpA
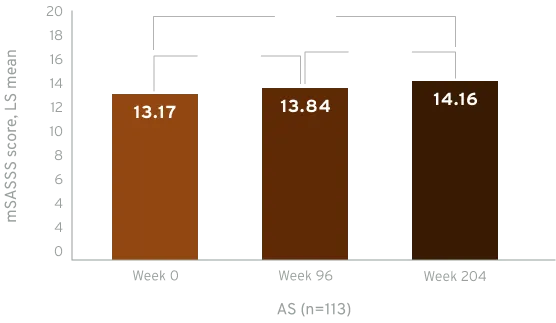
Active AS patients demonstrated limited spinal radiographic progression over 4 years, with decreasing progression from year 2 to year 4 (week 96 to week 204)6jkl
Image
Image
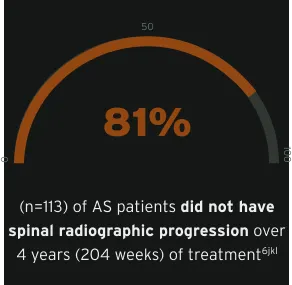
Radiographic data were examined for all CIMZIA®-treated patients with ≥1 mSASSS assessment (X-ray set), including those rerandomised from placebo. Based on average scores of the two readers, LS mean mSASSS and changes between visits were estimated using MMRM analyses on observed data.6
In the RAPID-axSpA study, the primary endpoint was met. The primary endpoint was ASAS20 at week 12. ASAS20 response rates were significantly higher at week 12 for CIMZIA® 200 mg Q2W (n=111) and CIMZIA® 400 mg Q4W (n=107) arms vs. placebo (n=107) (57.7% and 63.6%, respectively, vs. 38.3%), p≤0.004.15
RAPID-axSpA
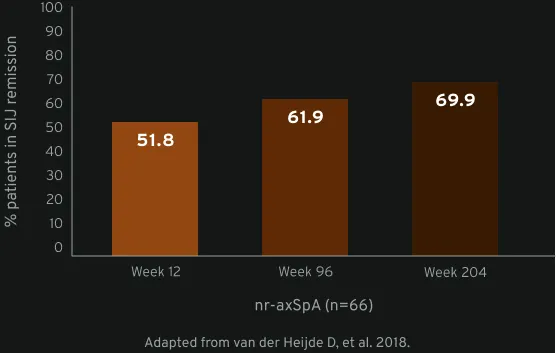
MRI SIJ remission (SPARCC score <2) achieved at week 12 was maintained until week 204 in active nr-axSpA6
Image
Image
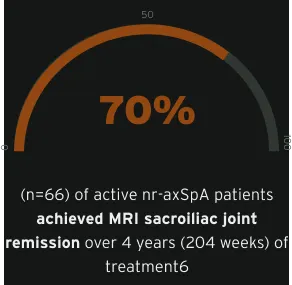
The MRI set included all randomised patients with valid MRI assessments (either spine or SIJ) at baseline and ≥1 other timepoint during the trial (n=158). Week 12 MRI data were not used from patients randomised to placebo. Average MRI scores of the two readers were considered for statistical analyses, and group LS mean Berlin and SPARCC scores were estimated post hoc by MMRM analysis on observed data using ‘visit’ as a fixed factor with an unstructured within-patient covariance matrix. The proportions achieving MRI remission (SPARCC <2 or Berlin ≤2) were estimated by multiple imputation: estimated proportions of patients in MRI remission were pooled from 50 multiply imputed data sets, where missing actual scores were imputed via predicted mean matching, with the predicted value at a visit based on linear regression of values from other visits. Results were summarised for patients with MRI baseline inflammation (Berlin score >2 or SPARCC score ≥2).6
RAPID-axSpA
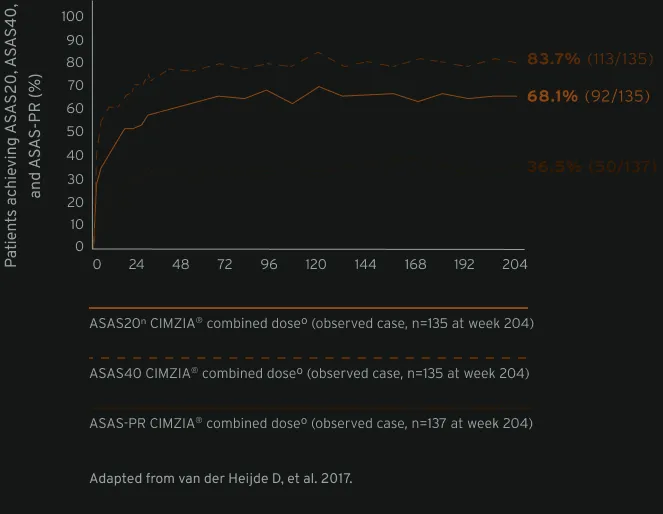
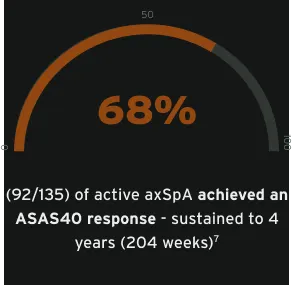
With long-term sustained clinical improvements (ASAS20, ASAS40, ASAS-PR), CIMZIA® is a treatment that appropriate women with active axSpA can consider at any stage of their life1,7m
CIMZIA® should only be used during pregnancy if clinically needed.1
Data from male and female patients.7
Image
Image
Extra-articular manifestations
How does CIMZIA® impact extra-articular manifestations in axSpA?
CIMZIA® is not registered for the treatment of uveitis or enthesitis.1
Please refer to the CIMZIA® SmPC for a complete list of licensed indications.1
Patient outcomes
Looking for patient outcomes in axSpA?
To find out how CIMZIA® impacts patient outcomes in axSpA, please connect with your UCB representative.
CIMZIA® safety profile
The use of adequate contraception should be considered for women of childbearing potential. For women planning pregnancy, continued contraception may be considered for 5 months after the last CIMZIA® dose due to its elimination rate, but the need for treatment of the woman should also be taken into account. Data from more than 1,300 prospectively collected pregnancies exposed to CIMZIA® with known pregnancy outcomes, including more than 1,000 pregnancies exposed during the first trimester, does not indicate a malformative effect of CIMZIA®. Further data are being collected as the available clinical experience is still limited to conclude that there is no increased risk associated with CIMZIA® administration during pregnancy. CIMZIA® should only be used during pregnancy if clinically needed. CIMZIA® can be used during breastfeeding.1
You may also like…
Image

Name of the page
Image

Name of the page
EU-P-CZ-PsA-2200001
Date of preparation: February 2023